Médica neurologista e neurofisiologista do Complexo Hospitalar Santa Casa de Porto
Alegre e do Hospital Moinhos de Vento. Título de Especialista pela Academia Brasileira de Neurologia e pela Sociedade Brasileira de Neurofisiologia Clínica (SBNC). Mestre em Clínica Médica pela UFRGS.
Alegre e do Hospital Moinhos de Vento. Título de Especialista pela Academia Brasileira de Neurologia e pela Sociedade Brasileira de Neurofisiologia Clínica (SBNC). Mestre em Clínica Médica pela UFRGS.
Francisco Tellechea Rotta
Médico neurologista. Coordenador do Ambulatório de Doenças Neuromusculares da Santa Casa de Misericórdia de Porto Alegre. Especialista em Neurologia e Doenças Neuromusculares pela University of Miami School of Medicine. Título de Especialista pela SBNC.
Fonte: http://www.medicinanet.com.br
Introdução
As doenças neuromusculares compreendem um grupo de doenças que direta (via distúrbio intrínseco do músculo) ou indiretamente (via distúrbio do neurônio motor, seus prolongamentos ou da junção neuromuscular) afetam a função muscular (Fig. 95.2).
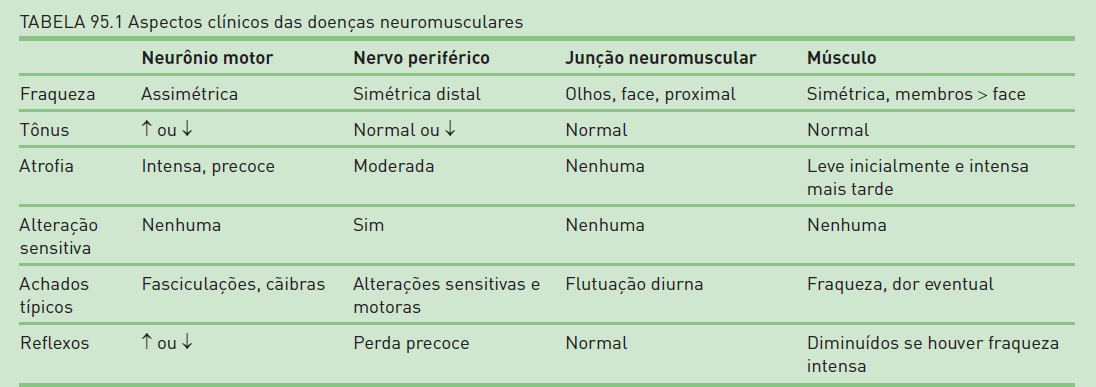
O diagnóstico é estabelecido basicamente por meio da história e do exame físico. Algumas informações são importantes para que o nível da lesão seja definido e para que seja realizada diferenciação entre as diversas doenças neuromusculares existentes (Tab. 95.1 e Quadro 95.1). A rapidez com que a doença progride (aguda, subaguda ou crônica), o padrão de acometimento dos sintomas (proximal, distal, face), a história familiar (forma hereditária ou adquirida) e os fatores desencadeantes também auxiliam na abordagem desses pacientes.


Doenças do Neurônio Motor
Esclerose lateral amiotrófica
Definição
A esclerose lateral amiotrófica (ELA) é uma doença neuro-degenerativa do sistema motor com comprometimento dos neurônios do corno anterior da medula e dos neurônios motores do córtex cerebral. Caracteriza-se pela combinação de sinais do neurônio motor superior e inferior.
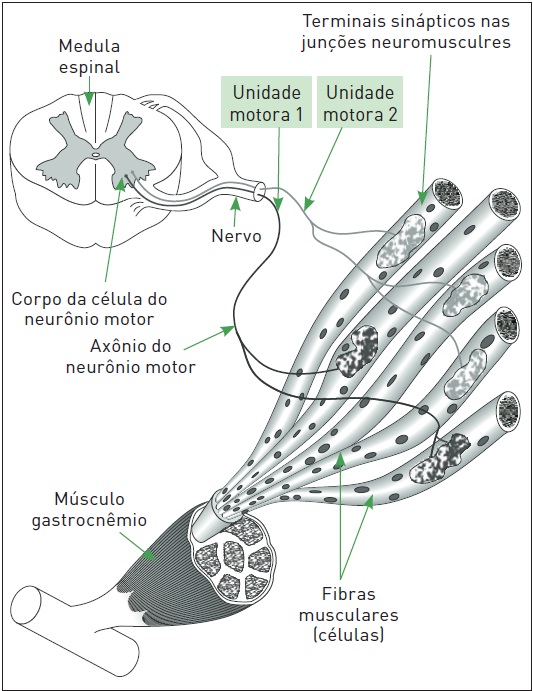
Figura 95.2
Via neurológica e muscular nas doenças neuromusculares.
Etiologia
A etiologia dessa doença é complexa e multifatorial. Geralmente é esporádica, mas pode ser familiar (5 a 10%) com herança autossômica dominante (gene que codifica a enzima superóxido-dismutase [SOD1]). Relaciona-se com a existência de algum fator genético, e a sua expressão clínica está relacionada com a exposição desse indivíduo, marcado geneticamente, a algum fator, ou fatores, que são desencadeadores do processo de degeneração do moto- neurônio (p. ex., processo inflamatório, envelhecimento, vírus, exposição a agentes tóxicos, etc.).
Epidemiologia
A incidência da ELA é rara, ocorrendo cerca de 2 casos para 100.000 pessoas/ano. Indivíduos do sexo masculino são mais afetados do que os do feminino em uma proporção de 2:1, e os brancos são mais acometidos do que os negros. A idade média de início é 50 anos, e a forma esporádica compreende mais de 90% dos casos. Entre os pacientes com a doença, 50% sobrevivem por três anos, e 28%, por mais de cinco anos.
Patogênese
Ao realizar exame macroscópico, nota-se um afilamento das raízes anteriores da medula. Por meio desse exame, torna-se evidente a perda neuronal no córtex motor, nos núcleos dos nervos cranianos e no corno anterior da medula, bem como a redução das fibras dos feixes corticobulbares e corticoespinais. É interessante que não haja evidência de processo inflamatório nas estruturas envolvidas.
Sinais e sintomas
Quando o paciente apresenta sinais proeminentes de envolvimento do neurônio motor superior e inferior nos dois membros e nos músculos bulbares (25% dos pacientes apresentam disartria, disfagia), sem comprometimento sensitivo, autonômico e visual, o diagnóstico clínico é evidente, e o diferencial é restrito.
Entretanto, o desafio encontra-se nos pacientes em estágio inicial da doença, que podem ter diferentes formas de apresentação clínica, dependendo do sítio acometido, alargando o diagnóstico diferencial (Tab. 95.2).
Diagnóstico
Até o momento, não há marcador biológico específico para pacientes com ELA, e o diagnóstico está baseado na história, no exame físico e em alguns achados de exames complementares, como os seguintes:
Eletroneuromiografia: comprometimento do neurônio motor em regiões clinicamente comprometidas e não comprometidas; denervação e reinervação (fasciculações, fibrilações, polifásicos com amplitude aumentada de unidades motoras) e velocidade de condução normal inicialmente.
Aspectos clínicos típicos de pacientes com esclerose lateral amiotrófica
Essa doença é caracterizada por fraqueza, sem comprometimento cognitivo, sensorial ou autonômico, e uma combinação de sinais dos neurônios motor superior (fraqueza, aumento do tônus, espasticidade, hiper-reflexia, cutâneo plantar em extensão, atrofia) e inferior (fraqueza, hipotonia, ausência de reflexos, atrofia e fasciculações).
Exames de neuroimagem: devem ser realizados a fim de descartar doenças que possam mimetizar a ELA.
Exames laboratoriais: devem ser realizados para descartar outras doenças ELA-like.

Tratamento
Para o tratamento de pacientes com ELA, utiliza-se riluzol (Rilutek®), 50 mg, duas vezes ao dia, administrado em períodos distantes das refeições. Esse medicamento diminui a progressão da doença.
Como suporte, pode-se realizar gastrostomia, ventilação não invasiva, controle da saliva, fisioterapia, fono- terapia e utilizar antiespasmódicos.
Doenças dos Nervos Periféricos
As doenças dos nervos periféricos formam um conjunto de síndromes clínicas que afetam nervos periféricos sensitivos, motores e/ou autonômicos. Apesar das diversas etiologias dessas doenças, as respostas a uma lesão são limitadas: axonopatias, neuronopatias ou mielinopatias. Na abordagem diagnóstica, a primeira etapa é determinar, por meio da história e exame físico, se o paciente apresenta uma neuropatia periférica (ver Quadro 95.2).
Determinada a existência de neuropatia, deve-se caracterizar a síndrome, levando em consideração alguns parâmetros, desde a rapidez com que se estabelece até a distribuição e o padrão dos achados clínicos. Dessa forma, pode-se limitar as possibilidades diagnósticas e planejar os exames complementares (ver Figura 95.3).
Rapidez de estabelecimento: aguda: horas até um mês; subaguda: um a dois meses; crônica: mais de dois meses.
Tipo de fibra nervosa envolvida: motora, sensitiva, autonômica ou mista.
Tamanhodafibranervosaenvolvida: grossa (fibras motoras, propriocepção e vibração), fina (dor, temperatura e sintomas autonômicos) ou mista.
Distribuição: proximal, distal, difusa.
Padrão: mononeuropatia, polineuropatia ou mononeuropatia múltipla.
Patologia: axonal, desmielinizante ou mista.

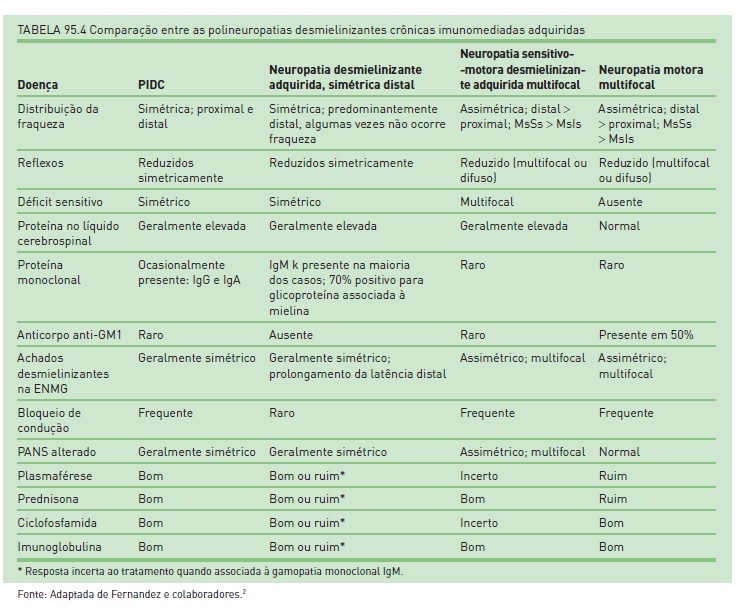
Síndrome de Guillain-Barré (polirradiculoneurite desmielinizante idiopática aguda [PDIA])
Definição
A síndrome de Guillain-Barré é uma doença desmielinizante que afeta os nervos periféricos, potencialmente grave, podendo resultar em falência respiratória aguda mesmo nos pacientes nos quais não há fraqueza dos membros.
Etiologia
Entre os pacientes com essa doença, 25% apresentam infecção prévia por Campylobacter jejuni, realizaram vacina, submeteram-se à cirurgia uma a três semanas antes do início dos sintomas, ou são mulheres grávidas.
Sinais e sintomas
Os pacientes com síndrome de Guillain-Barré apresentam fraqueza simétrica ascendente e rapidamente progressiva. Os reflexos tendinosos profundos ficam diminuídos ou podem inexistir. Podem ocorrer também diplegia facial (aspecto de máscara, ausência mímica facial), disfagia e dificuldade respiratória.
Diagnóstico
O diagnóstico dessa condição é clínico e confirmado por achados em exames complementares, tais como os seguintes:
Líquido cerebrospinal: proteína elevada depois de cinco a seis dias, celularidade normal.
Laboratorial: Campylobacter, HIV, CMV, FAN, FR, hepatites, GM1 E GQ1b anticorpos.
ENMG: bloqueio de condução, latência aumentada, diminuição da onda F.
Biópsia: desmielinizção focal (somente se houver dúvida quanto ao diagnóstico).
Respiratório: capacidade vital forçada três vezes ao dia.
A síndrome de Miller-Fischer é uma variante clínica caracterizada por arreflexia, ataxia sensitiva, oftalmoparesia e existência do anticorpo GQ1b.
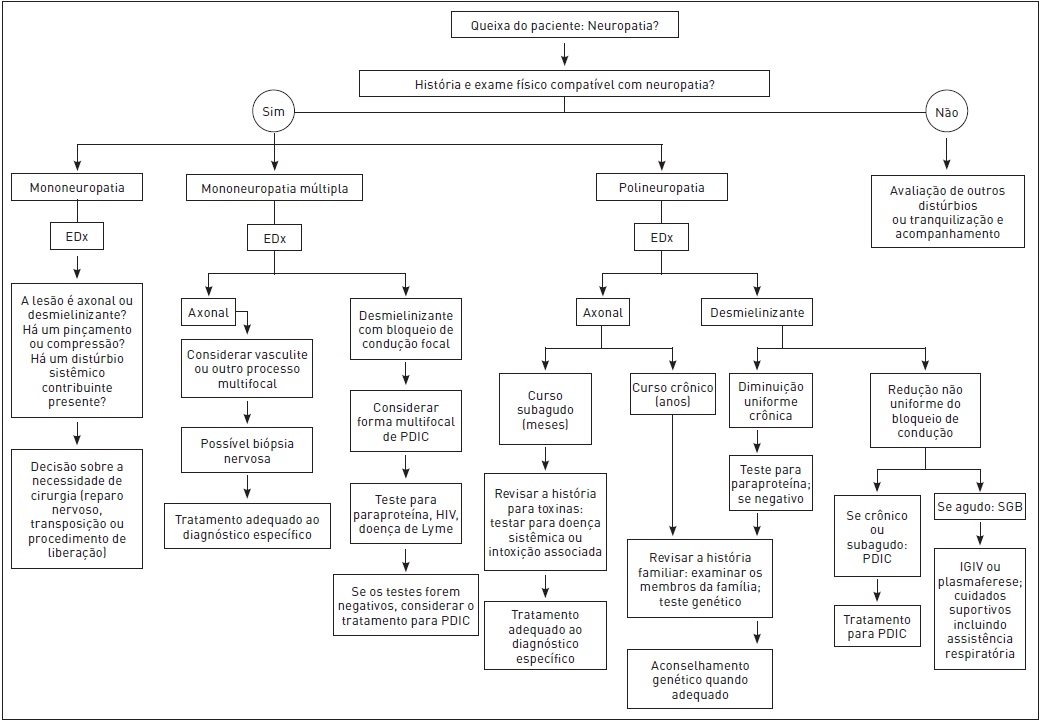
Figura 95.3
Abordagem à avaliação de neuropatias periféricas.
PDIC, polirradiculoneuropatia desmielinizante inflamatória crônica; EDx, estudos de eletrodiagnóstico; SGB, síndrome de Guillain-Barré; IGIV, imunoglobulina intravenosa.
Diagnóstico diferencial
Deve ser realizado diagnóstico diferencial dessa síndrome com polirradiculopatia inflamatória desmielizante crônica (PIDC), miastenia, botulismo, doença de Lyme, esclerose múltipla, tumores medulares, poliomielite e hipofosfatemia.
Tratamento
Para pacientes com essa doença, pode-se realizar ventilação mecânica se a capacidade vital for menor do que 15 cc/kg ou 0,6 L, ou maior, se estiver reduzindo rapidamente.
Outras opções igualmente efetivas são plasmaférese e imunoglobulina IV. A administração de corticoide não apresenta benefício.
Prognóstico
O prognóstico desses pacientes é fatal em 4 a 15%, deixa sequela em 20% e em 35% apresenta déficit permanente. Ocorre recaída em 10% dos indivíduos, 50% atingem o nadir em uma semana, e 90%, em um mês.
Polirradiculopatia inflamatória desmielinizante crônica (PIDC)
Definição
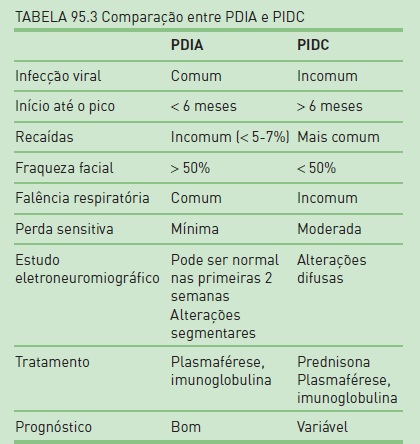
A PIDC é semelhante à PDIA em relação aos sintomas clínicos, exceto pelo curso remitente recorrente, com duração de, pelo menos, dois meses (Tabs. 95.3 e 95.4).
Doença de Charcot-Marie-Tooth (CMT)
Definição
A CMT é causada por mutações em genes que codificam proteínas de diferentes localizações (mielina, células de Schwan e axônios) e que estão envolvidas em diversas funções: desde compactação e manutenção da mielina para formação do citoesqueleto, transporte axonal até metabolismo mitocondrial. Independentemente desse processo, a lesão final que se estabelece é uma degeneração axonal, principalmente nas fibras grossas e longas.
Epidemiologia
A CMT é uma das doenças neuromusculares hereditárias mais comuns, com uma prevalência de mais de 40/100.000 pessoas.
Etiologia
Essa doença é geneticamente heterogênea com diferentes mutações resultando em fenótipo clínicos semelhantes. Também conhecida como neuropatia sensitivo-motora hereditária. A maioria das pessoas com CMT apresenta herança autossômica dominante, mas todos os tipos de transmissão podem ser observados.
Sinais e sintomas
O fenótipo típico da CMT é de atrofia, fraqueza e perda sensitiva com início distal e progressão proximal. Os sintomas motores manifestam-se inicialmente nos pés, com fraqueza na musculatura intrínseca, pés cavos e dedo em martelo, diminuição da sensibilidade vibratória, dor ao toque. Algumas vezes, ocorre perda da sensibilidade proprioceptiva, ocasionando ataxia sensitiva. Não há reflexos tendinosos profundos (Quadro 95.3).
Diagnóstico e classificação
Para iniciar a abordagem diagnóstica, deve-se definir o fenótipo clínico, identificar o padrão de herança, realizar ENMG, estudo molecular e, para casos selecionados, biópsia de nervo.
A classificação é estabelecida com base nos estudos de condução nervosa e na patologia do nervo. Em pacientes com CMT-1, verificam-se lentificação da velocidade de condução nos estudos neurofisiológicos e anormalidades da mielina na patologia. Já em indivíduos com CMT-2, há velocidades de condução normais ou pouco reduzidas e achados de degeneração axonal crônica e regeneração. As subdivisões dessa classificação são baseadas nos tipos de genes alterados.
Diagnóstico diferencial
Deve-se realizar diagnóstico diferencial da CMT com outras neuropatias hereditárias, adquiridas, miopatias distais, doença do neurônio motor, ataxias hereditárias, paraplegias espásticas hereditárias, doenças mitocondriais e leucodistrofias.

Tratamento
Ainda não há tratamento medicamentoso efetivo para pacientes com CMT. O tratamento de suporte consiste em fisioterapia, órteses e correções cirúrgicas das deformidades, sendo necessária uma abordagem multidisciplinar.
Neuropatia diabética
Definição
A neuropatia diabética é causada por diabetes melito (DM). Diversas síndromes clínicas foram identificadas, tais como as seguintes:
Mononeuropatias agudas
Mononeuropatias múltiplas e radiculopatias
Polineuropatia distal
Neuropatia autonômica
Epidemiologia
A neuropatia diabética é incomum antes dos 30 anos. Quinze por cento dos pacientes com DM tipo 1 e 13% dos pacientes com tipo 2 apresentam polineuropatia sintomática, mas as taxas de ocorrência aumentam quando se avalia a ENMG desses pacientes.
Patogênese
Perda de fibras nervosas mielinizadas é o achado mais proeminente em casos de polineuropatia distal. Desmielinização e remielinização segmentar, formação de bulbo com aspecto de cebola e redução do número de fibras não mielinizadas são achados secundários à isquemia aguda da vasa nervorum.
Diagnóstico
O diagnóstico dessa condição é clínico, e a realização de ENMG confirma-o.
Tratamento
Nesses casos, deve-se manter níveis glicêmicos adequados. Pode-se realizar tratamento sintomático com antidepressivos tricíclicos e anticonvulsivantes.
Neuropatia nutricional
Definição
A neuropatia nutricional é uma doença dos nervos periféricos que ocorre devido à deficiência isolada ou complexa de nutrientes. A causa mais comum dessa doença no Ocidente está associada ao uso de álcool.
Epidemiologia
Estima-se que entre 25 a 66% dos alcoolistas crônicos desenvolvam neuropatia durante a vida. Os fatores diretamente associados ao desenvolvimento de neuropatia são o período em que se consome o álcool e a quantidade.
Patogênese
A neuropatia nutricional é causada por lesão axonal de fibras nervosas de pequeno calibre associada às seguintes hipóteses distintas ou associadas:
Deficiência de tiamina: o etanol reduz a absorção intestinal de tiamina e também o estoque hepático e altera a fosforilação dela. A dieta do paciente com alcoolismo crônico geralmente é desequilibrada.
Efeito neurotóxico direto do etanol ou de seus metabólitos (acetaldeído): dose-dependente, alteração na modulação da cascata de sinalização intracelular da qual a proteína quinase A e C fazem parte; desequilíbrio do transporte axonal e das propriedades do citoesqueleto.
Sinais e sintomas
Pacientes com neuropatia alcoólica pura (sem evidência de deficiência de tiamina) apresentam sintomas sensitivos (diminuição da sensibilidade superficial e dor) lentamente progressivos, que se manifestam de forma inicial nos membros inferiores. Também podem posteriormente apresentar fraqueza, queimação, ataxia e quedas.
Quando associada a déficits nutricionais, a apresentação clínica é variável.
Diagnóstico
O diagnóstico pode ser determinado por meio das seguintes avaliações:
História de uso de álcool (100 mL de álcool etílico por dia durante três anos)
ENMG: não é específica para neuropatia alcoólica; polineuropatia sensitivo-motora axonal.
Exames laboratoriais para exclusão de outras doenças: eletrólitos, função renal, função hepática, glicose, funções da tireoide, níveis de tiamina, piridoxina, vitamina B12, ácido fólico, niacina, vitamina A, biotina; metais pesados, VSG, HIV, sífilis.
Biópsia de nervo.
Avaliação do tubo gastrintestinal.
Diagnóstico diferencial
Deve-se realizar o diagnóstico diferencial de neuropatia nutricional com esclerose lateral amiotrófica, deficiência de tiamina, doença de Charcot-Marie-Tooth, neuropatia diabética, mononeurite múltipla, neuropatia tóxica por metal pesado, neuropatias paraneoplásicas.
Tratamento
O tratamento de pacientes com neuropatia nutricional objetiva controlar os sintomas, treinar a capacidade de funcionamento independente, prevenir lesões, cessar o consumo de álcool, administrar suplementação vitamínica e calórica e medicações para dor neuropática e realizar fisioterapia.
Neuropatias por vasculites
Definição
As neuropatias por vasculites são um grupo heterogêneo de doenças que afetam variados órgãos e vasos sanguíneos de diferentes diâmetros.
Sinais e sintomas
São sinais e sintomas de pacientes com neuropatias por vasculites: mononeuropatia dolorosa, polineuropatia assimétrica ou mononeuropatia múltipla de início agudo ou subagudo. Elas afetam principalmente os membros inferiores. Os sintomas sistêmicos são mialgias, artral- gias, perda de peso, sintomas respiratórios, dor adominal, hematúria, rash e sudorese noturna.
Classificação
As neuropatias por vasculites podem ser classificadas das seguintes formas:
Sistêmicas:
–Primárias: sem causa definida; poliarterite nodosa, granulomatose de Wegener, síndrome de Churg- Strauss e poliangeíte microscópica.
–Secundárias: drogas, vírus, doenças do tecido conectivo; artrite reumatoide, lúpus eritematoso sistêmico, crioglobulinemia associada ao vírus C, HIV, citomegalovírus, sarcoidose.
Não sistêmica: restrita aos nervos e aos músculos.
Diagnóstico
O diagnóstico pode ser estabelecido por meio das seguintes avaliações:
ENMG: padrão axonal agudo ou subagudo, com envolvimento sensitivo e motor multifocal.
Laboratorial: hemograma, eletrólitos, ureia, creatinina, glicose, VSG, proteína Creativa, FAN, fator reumatoide (FR), anticorpo anticitoplasma de neutrófilos (ANCA), hepatite C e B, crioglobulinas, proteinogra ma, HIV, sífilis.
Biópsia de nervo e músculo: 60% apresentam deposição de imunocomplexos, inflamação e destruição da parede dos vasos.
Tratamento
Para o tratamento de pacientes que apresentam neuropatia com envolvimento sistêmico primário, deve-se administrar corticoide intravenoso (indução) e imunossupressores (manutenção). Para os casos de neuropatia com envolvimento sistêmico secundário, é contraindicado o uso de imunossupressão crônica, e o tratamento é direcionado para a doença de base.
Devido à benignidade da neuropatia não sistêmica, geralmente não é realizado tratamento para esses pacientes. Pode-se utilizar corticoide via oral ou, se a doença for mais agressiva, imunossupressores.
Doenças da Junção Neuromuscular
Miastenia grave
Definição
A miastemia grave é uma doença autoimune potencialmente grave, mas que pode ser tratada. Caracteriza-se por fraqueza e fatigabilidade da musculatura voluntária.
Epidemiologia
A incidência anual dessa doença é de 2/100.000 pessoas/ ano, mas a prevalência pode chegar a mais de 50/100.000 pessoas. Ela apresenta dois picos de incidência, o primeiro entre 20 e 30 anos, afetando mais mulheres, e o segundo entre 60 e 70 anos, acometendo mais homens.
Etiologia e patogênese
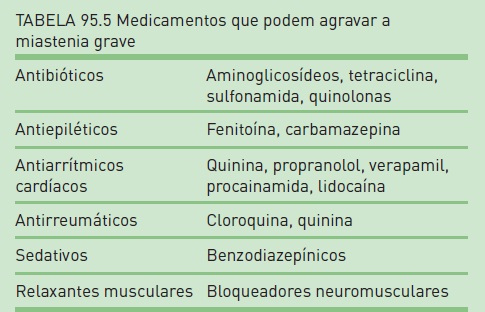
Os fatores que desencadeiam ou induzem o processo autoimune ainda não são conhecidos. Na maior parte dos casos, o alvo do ataque autoimune é o receptor nicotínico de acetilcolina (85% dos casos), localizado na membrana pós-sináptica da placa mioneural. Metade dos pacientes soronegativos apresenta um anticorpo antiquinase muscular específica (anti-MuSK). A perda de um grande número de receptores de acetilcolina nos músculos limita a quantidade de fibras que se despolarizam durante a ativação do músculo pela terminação nervosa, diminuindo, assim, a geração de ações musculares e impedindo a contração das fibras.
Os principais mediadores da resposta autoimune em casos de miastemia grave são os linfócitos T derivados do timo. No exame anatomopatológico, é possível verificar infiltrado linfocítico associado a pequenos focos necróticos na fibra muscular, atrofia das fibras tipo 1 e 2, hiperplasiacomfolículos linfoides, evidenciando centros germinativos em 70% dos timos, e timoma em 10%.
Alguns fatores podem agravar a miastenia grave, tais como infeções e o uso de drogas (Tab. 95.5).
Sinais e sintomas
Nos casos de miastenia grave, cinco subgrupos de sinais e sintomas são conhecidos:
Grupo 1: comprometimento da musculatura ocular isolada.
Grupo 2: fraqueza generalizada leve.
Grupo 3: fraqueza generalizada moderada ou fraqueza bulbo-ocular leve a moderada.
Grupo 4: fraqueza generalizada ou bulbo-ocular grave.
Grupo 5: crise miastênica.
Entre os pacientes com miastenia ocular primária, 85 a 90% desenvolvem fraqueza generalizada nos dois anos seguintes. A forma mais comum é a generalizada, que se apresenta com fraqueza flutuante, havendo piora no final do dia e cansaço fácil na musculatura extraocular, bulbar e de extremidades. Alguns sinais e sintomas típicos são voz anasalada, dificuldade de mastigação, diplopia, ptose e paresia ocular. Os reflexos pupilares são poupados.
Diagnóstico
O diagnóstico pode ser estabelecido por meio das seguintes avaliações:
Farmacológica: teste do Tensilon® (edrofônio), droga anticolinesterásica intravenosa de curta ação (2 a 4 minutos).
Sorológica: anticorpo antirreceptor de acetilcolina, anticorpo antirreceptor muscarínico específico.
Eletrofisiológica: redução da amplitude do potencial de ação composto evocado durante a estimulação repetitiva supramáxima de um nervo. A ENMG de fibra única é mais sensível.
Imagem: raio X de tórax pode evidenciar massa mediastinal. A tomografia computadorizada (TC) de tórax deve ser realizada em todos os pacientes recém-diagnosticados.
Diagnóstico diferencial
Deve-se realizar o diagnóstico diferencial da miastenia grave com as seguintes doenças:
Queixa de cansaço fácil: histeria, depressão, neurose, síndrome da fadiga crônica.
Queixa ocular: miopatia mitocondrial, distrofia oculofaríngea, esclerose múltipla, paralisias isoladas de nervos cranianos.
Polimiosite, esclerose lateral amiotrófica, botulismo, tumores de tronco cerebral, síndrome de Guillain-Barré, PIDC, intoxicação por organofosforados, distrofia muscular, doenças da tireoide.
Tratamento
O tratamento pode ser realizado com a administração de anticolinesterásicos, corticosteroides, imunossupressores, imunoglobulina e com plasmaférese e timectomia.
Pacientes com crise miastênica (agravamento da mistenia em geral por infecção, drogas ou tratamento inadequado) e crise colinérgica (excesso de acetilcolina na junção neuromuscular devido ao uso excessivo de drogas anticolinesterásicas) apresentam fraqueza, e são crises consideradas emergências médicas pelo risco de insuficiência respiratória. Elas podem ocorrer concomitantemente (Tab. 95.6).
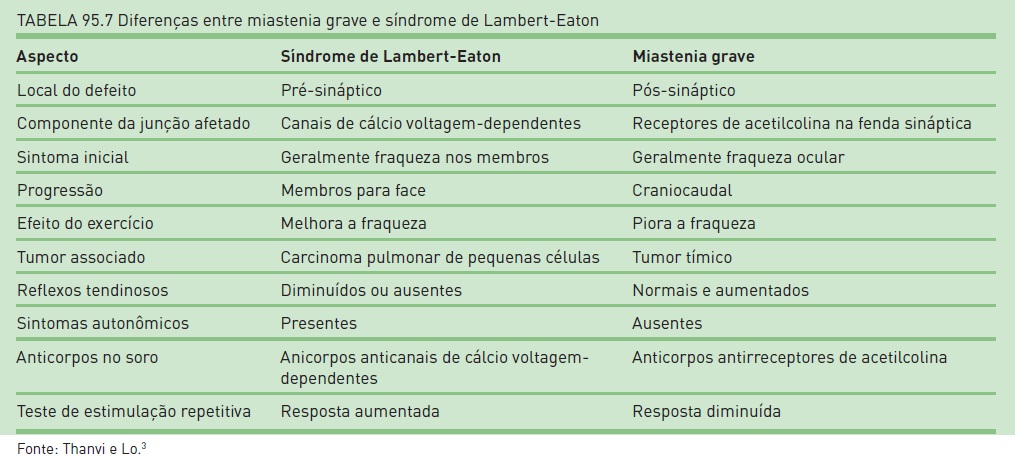
Síndrome miastênica de Lambert-Eaton
Definição
A síndrome miastênica de Lambert-Eaton é uma doença caracterizada por fraqueza generalizada (geralmente musculatura proximal) e por sinais e sintomas autonômicos (boca seca, constipação, impotência e inexistência de reflexos), que, ao contrário da miastenia grave, melhoram com exercício físico.

Etiologia e patogênese
Entre os pacientes com essa síndrome, 50% dos casos estão associados à neoplasia (geralmente neoplasia pulmonar de pequenas células) e, nestes, os anticorpos antiantígenos tumorais reagem com os canais de cálcio voltagem-depedentes envolvidos na liberação de acetil- colina na fenda pré-sináptica, ocasionando distúrbio na transmissão neuromuscular.
Diagnóstico
O diagnóstico é confirmado por ENMG por meio da respos ta à estimulação repetitiva. Ao contrário da miastenia grave, ocorre um aumento da resposta muscular ao estímulo repetitivo do seu nervo motor. A existência de anticorpos contra o subtipo P/Q dos canais de cálcio voltagem-dependentes é altamente sensível e específica para a síndrome, independentemente da etiologia (Tab. 95.7).
Tratamento
O tratamento é realizado com drogas imunosupressoras (corticoides, azatioprina), plasmaférese, imunoglobulina, 3,4-diaminopiridina. A administração de piridostigmina ou neostigmina não é efetiva como em casos de miastenia grave. Deve-se evitar drogas que afetam a junção neuromuscular.
Doenças do Músculo
Existem inúmeras doenças neuromusculares resultantes do comprometimento dos músculos estriados. Essa diversidade evidencia um caráter desafiador para o diagnóstico dessas doenças, já que elas apresentam um número limitado de sinais e sintomas: fraqueza, fadiga, dor, rigidez, espasmos, cãibras e alteração de volume, sem haver alteração da sensibilidade e com reflexos preservados inicialmente. A fraqueza muscular verdadeira é o sintoma mais frequente. A abordagem inicial do paciente com fraqueza deve levar em consideração o padrão topográfico de acometimento muscular (proximal, distal, facial), o período de aparecimento dos sintomas (agudo, subagudo ou crônico), o histório familiar (doença hereditária ou adquirida) e os sinais e sintomas associados.
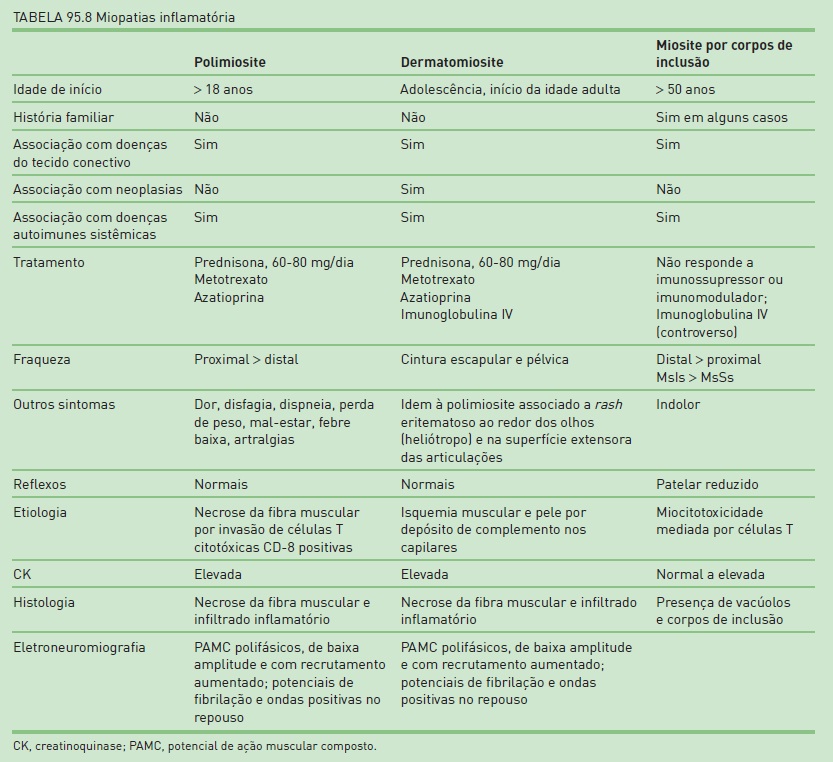
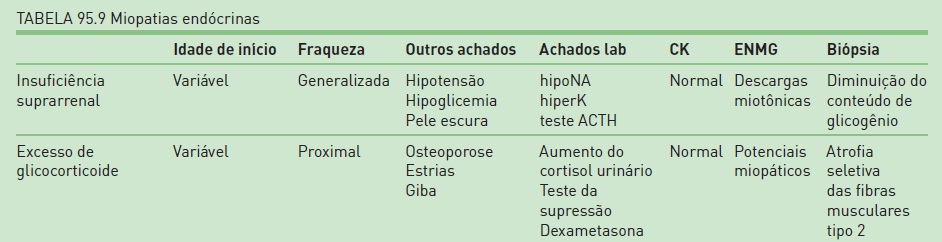
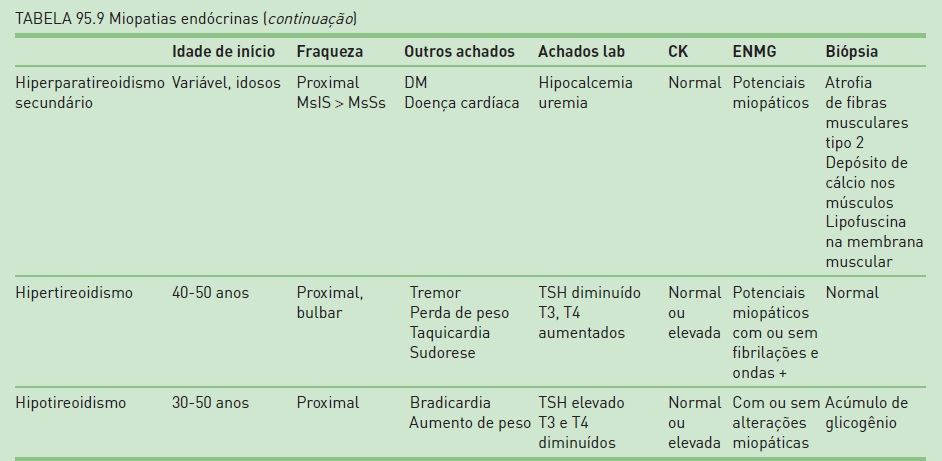
É possível classificar essas doenças em quatro grandes grupos: miopatias inflamatórias (infecciosas ou idiopáticas autoimunes [Tab. 95.8]), distrofias musculares (doenças degenerativas progressivas hereditárias), miopatias tóxicas e metabólicas (anormalidade metabólica hereditária, anormalidade da função endocrinológica [Tab. 95.9], drogas ou agentes miotóxicos) e miopatias congênitas. As miopatias inflamatórias constituem o grupo de doenças musculares mais comuns e potencialmente tratáveis.
Caso Clínico Comentado
A combinação de sinais do neurônio motor superior (fraqueza, aumento do tônus, espasticidade, hiper-reflexia, cutâneo plantar em extensão, atrofia) e inferior (fraqueza, hipotonia, ausência de reflexos, atrofia e fasciculações), a apresentação inicial assimétrica e os sinais bulbares (disartria, disfagia) evidenciam o diagnóstico de esclerose lateral amiotrófica. A rápida evolução desse paciente associa-se ao precoce envolvimento bulbar e à possibilidade de distúrbios respiratórios fatais.
Referências
1.Oliveira AS, Pereira RD. Amyotrophic Lateral Sclerosis (ALS): three letters that change the people’s life forever. Arq Neuropsiquiatr. 2009;67(3A):750-82.
2.Fernandez HH, Eisenschenk S, Okun MS. Ultimate review for the neurology boards. 2nd ed. New York: Demos; 2010.
3.Thanvi BR, Lo TC. Update on myasthenia gravis. Postgrad Med J. 2004;80(950):690-700.
4.Saguil A. Evaluation of the patient with muscle disease. Am Fam Physician. 2005;71(7):1327-36.
Flaherty AW, Rost NS, editors. The Massachusetts General Hospital handbook of neurology. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2007.
Koike H, Sobue G. Alcoholic neuropathy. Curr Opin Neurol. 2006;19(5):481-6.
Mammen AL. Dermatomyositis and polymyositis: clinical pre- sentation, autoantibodies and pathogenesis. Ann N Y Acad Sci. 2010;1184:134-53.
Pareyson D, Marchesi C. Diagnosis, natural history, and managment of Charcot-Marie-Tooth disease. Lancet Neurol. 2009;8(7):654-67.
Patten J. Diagnóstico diferencial em neurologia. 2. ed. Rio de Janeiro: Revinter; 2000.
Leituras Recomendadas
Aminoff MJ, Greenberg D, Simon RR. Clinical neurology. 6th ed. New York: McGraw-Hill; 2005.
Bloomgarden ZT. Diabetic neuropathy. Diabetes Care. 2007;30(4):1027-32.
Ropper AH, Brown RH. Adams and Victor´s: principles of neurology. 8th ed. New York: McGraw-Hill; 2005.
Schaublin GA, Michet CJ Jr, Dyck PJ, Burns TM. An update on yhe classification and treatment of vasculitic neuropahty. Lancet Neurol. 2005;4(12):853-65.